Oxokarbeniový ion
Oxokarbeniový ion je karbokation s sp2-hybridizovaným centrálním atomem uhlíku a navázaným atomem kyslíku.[1] Tyto ionty mají dvě rezonanční struktury, kde jednu představuje karbeniový ion s kladným nábojem na uhlíku a druhou oxoniový ion, s nábojem na kyslíku.
Oproti neutrálním karbonylovým sloučeninám, jako jsou ketony a estery, má větší podíl na celkové struktuře karbeniový ion. Oxokarbeniové ionty jsou častými reaktivními meziprodukty hydrolýz glykosidových vazeb a využívají se při chemických glykosylacích. Objevují se také v mechanismech enzymatických syntéz i hydrolýz sacharidů.
Barviva antokyaniny obsahují flavyliové ionty, což jsou stabilizované ionty oxokarbeniové.
Rozdělení elektronů a reaktivita
Lewisovská struktura nejlépe popisující oxokarbeniový ion obsahuje dvojnou vazbu C-O, kde je kyslík navázán na další skupinu a získává tak kladný náboj. Polarizace vazby π se popisuje pomocí rezonance sekundárního karbokationtu, s kladným nábojem na uhlíku. Podle teorie hraničních molekulových orbitalů je LUMO oxokarbeniového iontu orbital π* s větším lalokem na atomu uhlíku; elektronegativnější kyslík má na LUMO menší podíl; z tohoto důvodu působí uhlík jako elektrofil. Oproti ketonům se struktura oxokarbeniových iontů více podobá „pravým“ karbokationtům, což se projevuje vyšší reaktivitou vůči nukleofilům. Při organických reakcích bývají ketony často aktivovány navázáním Lewisových nebo Brønstedových kyselin na kyslík za vzniku oxokarbeniových meziproduktů.
Vznik
Oxokarbeniové ionty se mohou vytvářet několika způsoby. Nejčastěji na sebe karbonylový kyslík naváže Lewisovu kyselinu, která zvýší jeho elektrofilitu; touto Lewisovou kyselinou může být atom vodíku, ale i komplexní sloučenina, nejčastěji je takto navázaným atomem uhlík.
Další možností je eliminační reakce, vedoucí k oddělení odstupující skupiny; u sacharidů je touto skupinou často ether nebo ester. Kromě eliminace může také proběhnout přímá deprotonace, která je ovšem obtížnější a objevuje se jen za přítomnosti silných zásad.

Reakce
U pětičlenných kruhů
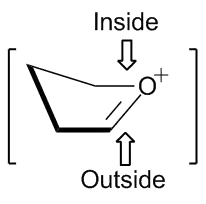
Stereochemii reakcí pětičlenných kruhů lze předvídat pomocí modelu obálkového přechodného stavu. Nukleofily se přednostně navazují z vnitřní strany obálky. Vnitřními adicemi vznikají produkty v nezákrytové, zatímco vnějšími adicemi v zákrytové konformaci.[2]
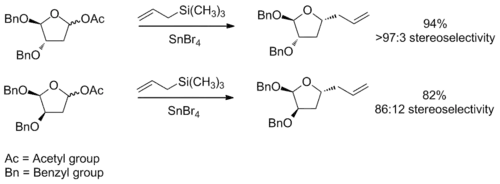
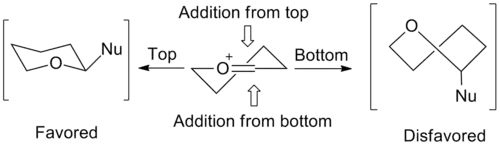
U šestičlenných kruhů
Model přechodného stavu u šestičlenného oxokarbeniového kruhu byl navržen v roce 1992.[3]
Stereochemie nukleofilních adicí na šestičlenné kruhy se určují podobně jako u pětičlenných kruhů. Základním předpokladem je přitom ten, že kruh je ve stejné konformaci jako cyklohexen, s třemi uhlíkovými atomy a jedním kyslíkem ve stejné rovině a dvěma zbylými uhlíky mimo ni, přičemž je jeden nad a jeden pod rovinou. Pode přítomných substituentů a jejich sterických a elektronových efektů lze určit konformaci s nejnižší energií; následně se uvažuje nukleofilní adice na molekulu s takovouto konformací. Adice proběhne skrz přechodný stav v lodičkové konformaci, která má nejnižší energii.
Na následujícím obrázku je uveden příklad takové reakce se znázorněním vlivu stereoelektronových efektů na elektronegativní substituent, kde získává nejnižší energii jiná konformace a obrací se selektivita.[4]
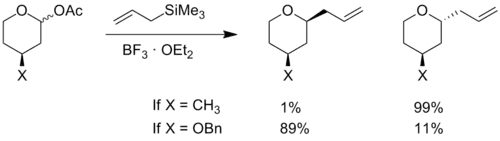
Stereoelektronové efekty
Na cykloalkenu neobsahujícím atom kyslíku budou větší substituenty navázány převážně do ekvatoriální pozice, kdy má molekula nejslabší sterické efekty, kruhy obsahující oxokarbeniové ionty ale mívají elektronegativní substituenty v axiálním nebo pseudoaxiálním uspořádání. Pokud je elektronegativní atom v axiální poloze, tak může dodávat elektronovou hustotu na kladně nabitý kyslík v kruhu,[5] čímž se stabilizuje axiální uspořádání; uvedený jev se vyskytuje například u hydroxylových, etherových, a halogenidových skupin. Při určování konformace s nejnižší energií je třeba také brát v úvahu stereoelektronové efekty.[6]

Cykloadice
V organické syntéze lze vinyloxokarbeniové ionty (struktura vlevo) využít k cykloadičním reakcím - tyto ionty jsou například častými dienofily v Dielsových–Alderových reakcích. K dienofilům se někdy za účelem urychlení reakce přidávají ketony se skupinami odtahujícími elektrony,[7] které se obvykle během reakce přeměňují na vinyloxokarbeniové ionty.[8]
Není známo, zda se oxokarbeniový ion vytvoří vždy, ale v níže zobrazené reakci byl potvrzen; reakce měla dva různé produkty, což je možné vysvětlit pouze přítomností oxokarbeniového kruhu u meziproduktu.[9] Popsány jsou rovněž [4+3], [2+2], [3+2], a [5+2] cykloadice zahrnující oxokarbeniové meziprodukty.[8]

Aldolové reakce
Chirální oxokarbeniové ionty lze využít k vysoce diastereoselektivním a enantioselektivním acetátovým aldolovým adicím.[10] Oxokarbeniové ionty zde slouží jako elektrofily. Diastereoselektivita roste s velikostí substituentu.
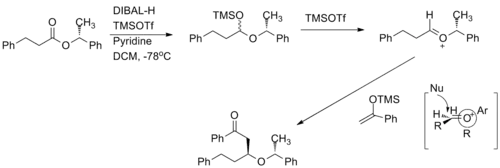
Příklady totálních syntéz
Oxokarbeniové ionty našly využití v několika totálních syntézách.
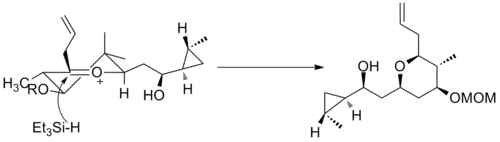
Jedna z podjednotek (+)-klavosolidu byla vytvořena redukcí šestičlenného oxokarbeniového kruhu. Všechny velké substituenty se nacházely v ekvatoriálních polohách a reakce probíhala, podle předpokladů, přes přechodný stav s židličkovou konformací.[11]
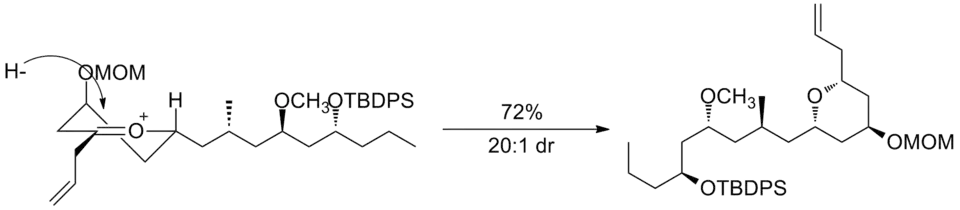
Druhým příkladem je jeden z kroků přípravy (−)-neopeltolidu, kde byla redukce jiného šestičlenného oxokarbeniového kruhu využita k diastereoselektivní adici hydridu.[12]
V biologii
V biologických systémech se oxokarbeniové ionty nejčastěji objevují v reakcích sacharidů. Protože jsou sacharidy (ribóza a deoxyribóza) součástmi nukleových kyselin, tak mají jejich reakce velký vliv na buněčné funkce nukleových kyselin. Sacharidy v organismech také slouží jako zásoba energie, signální molekuly, a v imunitním systému.[13]
Reakce nukleotidů
Nukleotidy mohou vstupovat do enzymatických vnitromolekulárních cyklizací a vytvářet tak několik biologicky významných molekul. Tyto cyklizace obvykle probíhají přes oxokarbeniové ionty. Příkladem látky vznikající těmito reakcemi je cyklická ADP ribóza, důležitá pro vnitrobuněčnou vápníkovou signalizaci.[14]
Glykosidázy
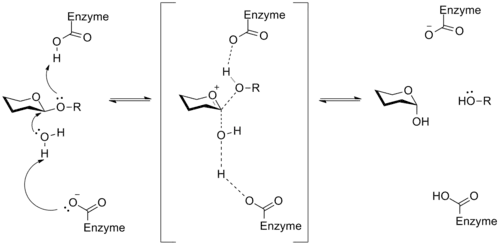
Glykosidázy jsou enzymy katalyzující rozklad glykosidových vazeb za vzniku dvou menších cukerných molekul. Tyto reakce, jako je štěpení glykogenu u živočichů a celulózy u rostlin, jsou důležitými součástmi energetického metabolismu; na katalytických místech příslušných enzymů se obvykle vyskytují zbytky kyseliny asparagové nebo glutamové. Mechanismus působení zahrnuje oxokarbeniové meziprodukty:[15]
Odkazy
Reference
V tomto článku byl použit překlad textu z článku Oxocarbenium na anglické Wikipedii.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. Příprava vydání Victor Gold. 4. vyd. Research Triangle Park, NC: International Union of Pure and Applied Chemistry (IUPAC) Dostupné online. doi:10.1351/goldbook.c00812. (anglicky) DOI: 10.1351/goldbook.
- ↑ C. H. Larsen; B. H. Ridgway; J. T. Shaw; K. A. Woerpel. A Stereoelectronic Model to Explain the Highly Stereoselective Reactions of Nucleophiles with Five-Membered-Ring Oxocarbenium Ions. Journal of the American Chemical Society. 1999, s. 12208–12209. doi:10.1021/ja993349z.
- ↑ R. J. Woods; C. W. Andrews; J. P. Bowen. Molecular mechanical investigations of the properties of oxocarbenium ions. 2. Application to glycoside hydrolysis. Journal of the American Chemical Society. 1992, s. 859–864. doi:10.1021/ja00029a008.
- ↑ J. A. C. Romero; S. A. Tabacco; K. A. Woerpel. Stereochemical Reversal of Nucleophilic Substitution Reactions Depending upon Substituent: Reactions of Heteroatom-Substituted Six-Membered-Ring Oxocarbenium Ions through Pseudoaxial Conformers. Journal of the American Chemical Society. 1999, s. 168–169. doi:10.1021/ja993366o.
- ↑ M. I. Miljkovic; D. Yeagley; P. Deslongchamps; Y. L. Dory. Experimental and Theoretical Evidence of Through-Space Electrostatic Stabilization of the Incipient Oxocarbenium Ion by an Axially Oriented Electronegative Substituent During Glycopyranoside Acetolysis. The Journal of Organic Chemistry. 1997, s. 7597–7604. doi:10.1021/jo970677d.
- ↑ Thomas Hansen, Ludivine Lebedel, Wouter A. Remmerswaal,Stefan van der Vorm, Dennis P. A. Wander, Mark Somers, Herman S. Overkleeft, Dmitri V. Filippov, Jérôme Désiré, Agnès Mingot, Yves Bleriot. Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Central Science. 2019-04-18, s. 781–788. ISSN 2374-7943. doi:10.1021/acscentsci.9b00042.
- ↑ Organic Chemistry. [s.l.]: [s.n.], 2009.
- ↑ a b {M. Harmata; P. Rashatasakhon. Cycloaddition reactions of vinyl oxocarbenium ions. Tetrahedron. 2003, s. 2371–2395. doi:10.1016/s0040-4020(03)00253-9.
- ↑ M. Roush; P. Gillis; A. Essenfeld. Hydrofluoric acid catalyzed intramolecular Diels–Alder reactions. Journal of Organic Chemistry. 1984, s. 4674–4682. doi:10.1021/jo00198a018.
- ↑ S. Kanwar; S. Trehan. Acetate aldol reactions of chiral oxocarbenium ions. Tetrahedron Letters. 2005, s. 1329–1332. doi:10.1016/j.tetlet.2004.12.116.
- ↑ J. D. Carrick; M. P. Jennings. An Efficient Formal Synthesis of (−)-Clavosolide a Featuring a "Mismatched" Stereoselective Oxocarbenium Reduction. Organic Letters. 2009, s. 769–772. doi:10.1021/ol8028302.
- ↑ D. Martinez-Solorio; M. P. Jennings. Formal Synthesis of (−)-Neopeltolide Featuring a Highly Stereoselective Oxocarbenium Formation/Reduction Sequence. The Journal of Organic Chemistry. 2010, s. 4095–4104. doi:10.1021/jo100443h.
- ↑ D. Anthea Maton; Jean Hopkins; Charles William McLaughlin; Susan Johnson; Maryanna Quon Warner; David LaHart; Jill D. Wright. Human Biology and Health. [s.l.]: Prentice Hall, 1993. Dostupné online. ISBN 0-13-981176-1. S. 52–59.
- ↑ H. M. Muller-Steffner; A. Augustin; F. Schuber. Mechanism of Cyclization of Pyridine Nucleotides by Bovine Spleen NAD+ Glycohydrolase. Journal of Biological Chemistry. 1996, s. 23967–23972. doi:10.1074/jbc.271.39.23967. PMID 8798630.
- ↑ D. L. Zechel; S. G. Withers. Glycosidase Mechanisms: Anatomy of a Finely Tuned Catalyst. Accounts of Chemical Research. 2000, s. 11–18. ISSN 0001-4842. doi:10.1021/ar970172. PMID 10639071.